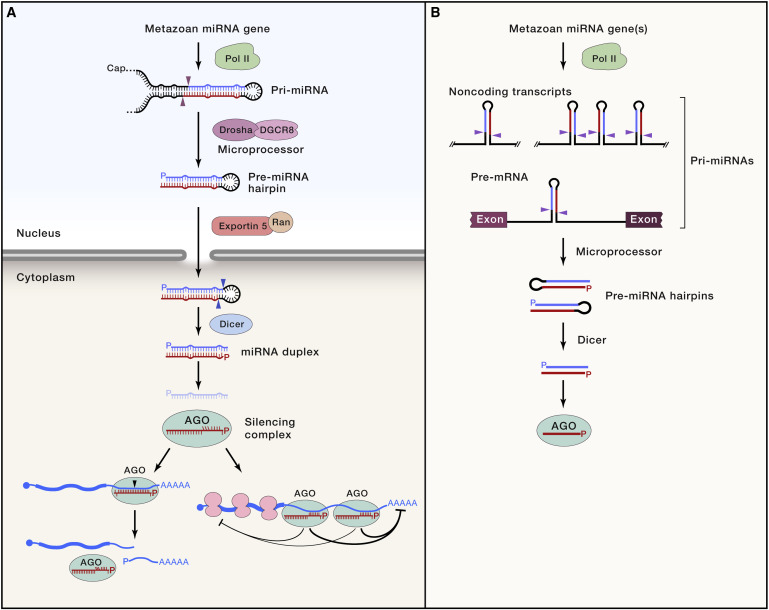
What are microRNAs?
MicroRNAs (miRNAs) form a class of small-non-coding RNAs that post-transcriptionally regulate mRNAs through RNA-induced silencing complex formation (RISC) and target mRNA binding. From each precursor miRNA two mature forms commonly arise (5p- major, 3p-major) one of which is selected to be degraded while the other is loaded into protein complexes assembled with AGO proteins to form RISCs. The canonical miRNA biogenesis involves transcription of miRNA genes, even though they may arise from several other origins such as intrions, also known as mirtrons.
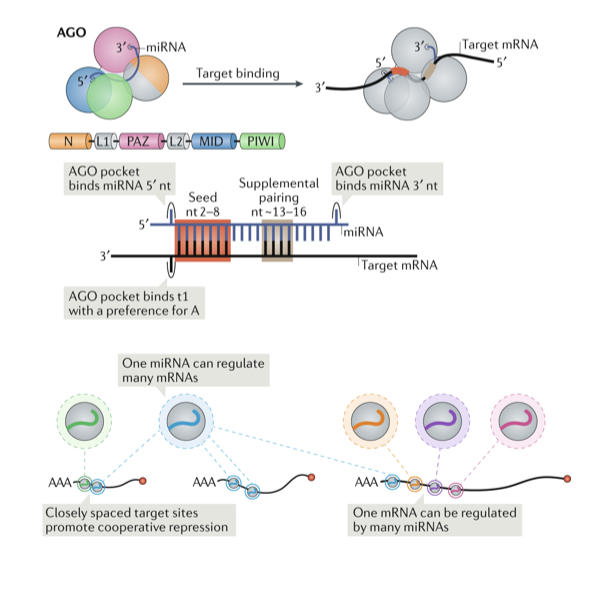
What is the function of microRNAs?
The canonical targeting mechanism involves binding of the RISC to the 3' untranslated region (UTR) of a target mRNA. This results in either repression of translation by blocking the ribosomal machinery, mRNA destabilization, or decay. Underlying the binding Watson-Crick basepairing is established between parts of the miRNA and the target sequence. Different types of sequence matches can influence the strength and efficacy of target regulation.
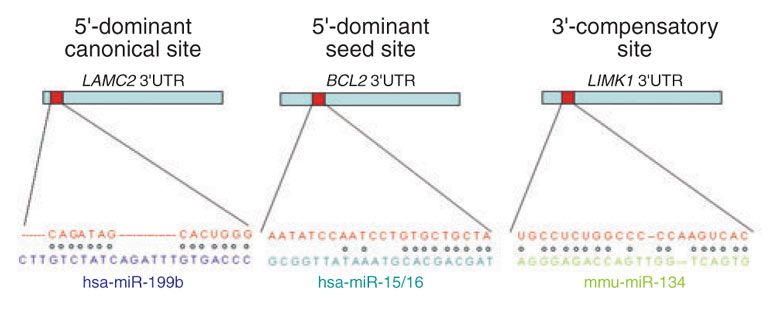
How do microRNAs target mRNAs?
The three most important types of miRNA-target sites defined by RNA Watson-Crick pairings are 5' -dominant canonical, 5'-dominant seed site, and 3'- compensatory site. Each site type involves a binding of the seed region that is the seed sequence defined in the mature miRNA from position 2 to 7 in 5' to 3' direction, to a target domain. The 3' supplementary bindings as shown for 5'-dominant canonical sites and 3' compensatory sites stabilize the bindings in case the seed region involves partial mismatches or G:U wobble pairs. In particular, the 3' compensatory binding can be specific for the seed region of certain miRNA family members, further enhancing the regulatory specificity.
What is a microRNA-target prediction tool?
In silico modelling of microRNA-target interactions promises several advantages. For example, one can analyse entire genomes for regulatory sites in a high-throughput manner and rank likely candidates for wet-lab validation. About 100 tools for this task have been developed so far, comprising a rich set of features, assumptions and input requirements. While early methods mostly were based on rules or probabilistic signal-to-noise considerations, later tools moved focus on data-driven approaches including machine and deep learning. The majority of tools either utilizes gene expression measurements, sequence information of miRNAs and mRNAs from genome assemblies, or both. Also, the tools generate several types of output each with its own advantages and disadvantages. We argue that prospective end-users working in life-science should be aware of the assumptions and output of microRNA-target prediction tools, that is why we implemented this website. The collection of tools is going to be updated as new publications in the field appear.
Which features can be exploited for target prediction?
Already hundreds of features have been proposed to characterise miRNA-target interactions. Most of it can be categorized into Sequence complementarity (SC), Structural & free energy properties (ST), Site accessibility and secondary structure (SA), Species conservation (C), and expression analysis (E). Over the time, additional features were proposed such as a preferential positioning of binding sites within UTRs, cooperativ targetting through clustering, or the composition of immediate neighbouring nucleotide content.
Why do some columns contain N/A values?
For some publications the respective criterion could not be determined using the publication material only. This may also be the case when the associated online resource (website, lab-page, etc.) was not reachable at the time the literature review was performed.
How did you compare the tools in your clustering?
The initial distance matrix underlying Figure 1 was computed using the gower distance implemented in the R package cluster and subsequently visualized with circlize, dendextend, and viridis. The 16 features selected for clustering the tools can be found in the next FAQ entry.
Which features were used for clustering the tools?
Please find below the list of features we used for the similarity analysis (Figure 1):
- Web server (Method)
- Download (Method)
- Download (Default predictions)
- Target organism
- Machine learning based
- Underlying method
- Used target principle(s)? Sequence compl. (SC), Structure (ST), Site Accessibility (SA), Conservation (C), Expression level (E)
- Requires NGS data for a user run
- Input data type? Sequence (S) / Expression (E) / None (N) / Both (B = S+E)
- Validation data type? Artificial (A) / Experimental (E) microRNA-target datasets
- Offers downstream analysis, e.g. pathway enrichments
- Uses predictions from other published tool(s), i.e. non ab initio
- Prediction granularity? Per binding site (BS) or per whole target (T) prediction.
- Target region? 5’ UTR, CDS, 3’ UTR, Any
- Main output type(s)
- Utility tool
Where can I download your data set?
You can acquire the data in several common formats using the dark-grey buttons at the top of the "Table" view, which is linked in the navigation bar.
Why is the PACCMIT-CDS tool categorised strangely?
The entry for PACCMIT-CDS represents the three tools PACMIT, PACCMIT and PACCMIT-CDS. These algorithms are similar in many aspects, so they were grouped together.
I have encountered an issue or bug. What to do next?
Please contact the authors under the following address and include a sufficient problem description: fabian.kern[at]ccb.uni-saarland.de
I have a question that is not covered here.
Please see the previous FAQ entry.
How can I cite this work?
Please cite the corresponding publication:
Kern, F. et al.
What’s the target: understanding two decades of in silico microRNA-target prediction. Brief Bioinform..
https://doi.org/10.1093/bib/bbz111 (2019).
Where can I get more information?
Please find below a list of publications that you may found useful, including technical benchmarks and in-depth review articles:
- Fan, X. & Kurgan, L.. Comprehensive overview and assessment of computational prediction of microRNA targets in animals. Briefings Bioinforma. https://doi.org/10.1093/bib/bbu044 (2014).
- Li, Y. et al. Systematic review of computational methods for identifying miRNA-mediated RNA-RNA crosstalk. Briefings Bioinforma. https://doi.org/10.1093/bib/bbx137 (2017).
- Tokar, T. et al. MirDIP 4.1 - Integrative database of human microRNA target predictions. Nucleic Acids Res. https://doi.org/10.1093/nar/gkx1144 (2018).
Please also consider the brief introduction texts in the other FAQ entries above.